Hereditary Cancers account for 5-10% of all cancers
Genetic Testing can aid in diagnosis, identify at-risk patients and improve outcomes
Inherited genetic mutations can increase a person’s risk of developing cancer, including gynecological cancers. The loss of cell growth control and the ability to repair damaged DNA is a common cause for different types of cancers. Variations in the genes involved in cell growth control and DNA repair are particularly likely to be associated with a general increased risk for cancer, resulting in the development of different cancers with the same genetic change(s) and overlapping clinical manifestations of multiple syndromic cancers. The genetic mutations that cause many of the known hereditary cancer syndromes have been identified. Genetic testing can confirm whether a condition is, indeed, the result of an inherited syndrome, reveal the causal genetic variation(s), assess cancer risk of a patient’s blood-related family members, and improve outcomes.
Otogenetics Comprehensive Panel addresses the overlapping genes found in multiple types of hereditary cancers. The utility of Next Generation Sequencing, the advanced genetic testing technology, allows the best possible coverage and detection of genetic variations associated with cancers and cancer risk.
Society of Gynecologic Oncology (SGO) Clinical Practice Statement on Next Generation Cancer Gene Panels Versus Gene by Gene Testing
“Advantages of cancer gene panels include decreased cost and improved efficiency of cancer genetic testing by decreasing the time involved, number of patient visits, and number of tests sent. A negative genetic test is more reassuring at eliminating the likelihood of inherited risk when all known genes for that phenotype have been assayed.”
“Approximately 15% of all ovarian cancers are attributable to a BRCA1 or BRCA2 mutation.
An additional 5-6% of ovarian cancers have gene mutations in other genes including the Lynch syndrome genes
(MLH1, MSH2, MSH6, PMS2), and BRIP1, RAD51D, RAD51C, PALB2, BARD1, and TP53”.
Indeed, data from 40,000+ hereditary cancer gene tests at Otogenetics show that additional genes contribute to more than 80% of the positive cases, while supporting the contribution from BRCA1 and BRCA2. Multi-gene testing allows for the identification of 40 to 50 percent more individuals with hereditary cancer gene mutations than is possible testing for BRCA1 and BRCA2 alone. The simultaneous evaluation of multiple genes is a highly cost-effective and time-effective option. In addition, some medical insurance allows coverage of genetic testing for a particular condition only once in a lifetime.
Otogenetics GxVision Hereditary Cancer Risk Assessment Tests
Gene Analysis Test Types
Otogenetics GxVision Hereditary Cancer Risk Assessment Test Options
Comprehensive Inherited Cancer Gene Tests – 45 genes
linked to Breast, Ovarian, Endometrial, Colorectal, Lynch Syndrome, Gastric, Melanoma, Pancreatic, Polyposis, Prostate, Renal, Thyroid/Parathyroid, Uterine and other major cancers
APC, ATM, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKn2A, CEBPA, CHEK2, DICER1, EPCAM, FANCC, GREM1, HOXB13, MEN1, MET, MLH1, MRE11A, MSH2, MSH3, MSH6, MUTYH, NBN, NF1, PALB2, PDGFRA, PMS2, POLD1, POLE, PTCH, PTEN, RAD50, RAD51C, RAD51D, RB1, RET, SMAD4, SMARCA4, STK11, TP53, VHL
Lynch Syndrome Genes – 5 genes
Breast, Ovarian and Uterine Cancer Genes or Gynecological Cancer Genes– 26 genes
ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CDKn2A, CHEK2, DICER1, EPCAM, MLH1, MSH2, MSH3, MSH6, MUTYH, NBN, NF1, PALB2, PMS2, PTEN, RAD50, RAD51C, RAD51D, SMARCA4, STK11, TP53
BRCA1/2 Genes – 2 genes
Colorectal Cancer Genes – 20 genes
APC, ATM, BMPR1A, BRCA1, CDH1, CHEK2, EPCAM, GREM1, MLH1, MSH2, MSH3, MSH6, MUTYH, PMS2, POLD1, POLE, PTEN, SMAD4, STK11, TP53
Prostate Cancer Genes – 13 genes
BRCA1, BRCA2, CDH1, CHEK2, EPCAM, HOXB13, MLH1, MSH2, MSH6, NBN, PMS2, PTEN, TP53
Melanoma Genes – 9 genes
ATM, BAP1, BRCA2, CDK4, CDKn2A, NBN, PTEN, RB1, TP53
Otogenetics Test Advantages
Accreditation Tests performed in CAP-accredited Next Generation Sequencing (NGS) laboratory in the US
Technology Robust and clinically actionable testing by the most advanced NGS technology
Quality Rare and common variants detected by high depth and complete sequence coverage
Reliable Reporting based on comprehensive external clinical genetic databases and an internal >100,000 patient database
Experience Among the first NGS laboratories for clinical testing and R&D
Guidelines from Professional Societies
American College of Obstetrics & Gynecology
ACOG Committee Opinion Number 634, June 2015
A hereditary cancer risk assessment is the key to identifying patients and families who may be at increased risk of developing certain types of cancer.
If a hereditary cancer risk assessment suggests an increased risk of a hereditary cancer syndrome, referral to a specialist in cancer genetics or a health care provider with expertise in genetics is recommended for expanded gathering of family history information, risk assessment, education, and counseling, which may lead to genetic testing.
American College of Medical Genetics & Genomics, ACMGG Practice Guidelines (doi:10.1038/gim.2014.147)
Hereditary breast–ovarian cancer (HBOC) syndrome is caused by mutations in BRCA1 and BRCA2 genes and is characterized by increased risks for early-onset breast, multiple breast primaries, male breast, and epithelial ovarian, Fallopian tube, or primary peritoneal cancers.
Cancers of the pancreas, prostate, and melanoma are more common in individuals with HBOC syndrome.
The pathology of “triple-negative phenotype” breast cancer (estrogen receptor–negative, progesterone receptor–negative, and HER2/neu–negative) has been strongly associated with BRCA1 mutations.
The likelihood of identifying a BRCA1/2 mutation in a woman with ovarian cancer at any age is around 13–18%.
Of males with breast cancer, 15–20% has a BRCA1/2 mutation.
The overall prevalence of BRCA1 mutations is estimated at 1 in 300. BRCA2 mutations are estimated at 1 in 800. Founder mutations in populations e.g., Ashkenazi Jewish, Icelandic, and Mexican Hispanic populations lead to increased mutation prevalence in these populations.
National Comprehensive Cancer Network, NCCN Clinical Practice Guidelines in Oncology
NCCN, Version 2.2021- November 20, 2020
Genetic testing should be considered for appropriate high-risk individuals where it will impact medical management of the tested individual and/or their at-risk family members.
The probability of a mutation being detected based on these criteria will vary depending family structure.
Individuals with unknown or limited family history, such as fewer than 2 female first- or second-degree relatives having lived beyond 45 years in either lineage, may have an underestimated probability of familial mutation detection.
The likelihood of detecting a mutation may be very low in families with a large number of unaffected female relatives.
Patients who have received an allogeneic bone marrow transplant should not have molecular genetic testing with current technologies using blood samples due to unreliable test results from contamination by donor DNA. DNA extracted from a fibroblast culture can be used. If not available, then a buccal sample can be considered with understanding of the risk of donor DNA contamination.
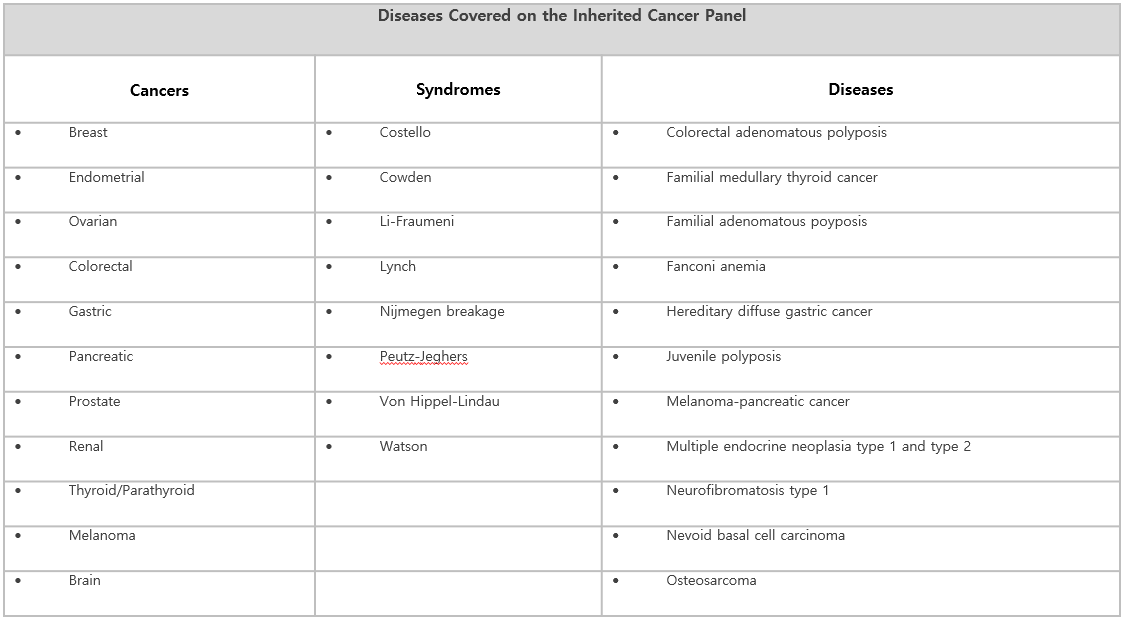
Diseases Associated With The Comprehensive Cancer Risk Assessment – 45 Gene Test
Interpretation of Test Results – Variant Classification
Variant classification is based on a Mendelian perspective and employs the recommended five-tier classification system recommended by the American College of Medical Genetics, ACMGG.
A sequence change can be classified as:
Positive – Pathogenic Variant(s) Detected
This result means that a mutation or a genetic change that increases the lifetime chance of developing certain cancers was identified. This variant directly contributes to the risk of developing cancer. Some pathogenic variants may not be fully penetrant. In the case of recessive conditions, a single pathogenic variant may not be sufficient to cause disease on its own. Additional evidence is not expected to alter the classification of this variant. The final report contains detailed risk information specific to the mutation(s). This result does not mean that the patient has cancer or will definitely develop cancer in their lifetime. Follow up with physicians, healthcare providers and genetic counseling is recommended.
Likely Pathogenic Variants(s) Detected
The result means that a mutation or a genetic change that is likely to increase the lifetime chance of developing certain cancers was identified. However current scientific evidence is insufficient to prove this conclusively. Additional evidence is expected to confirm this assertion of pathogenicity, but we cannot fully rule out the possibility that new evidence may demonstrate that this variant has little or no clinical significance. The final report contains detailed risk information specific to the mutation identified. This result does not mean that the patient has cancer or will definitely develop cancer in their lifetime. Follow up with physicians, healthcare providers and genetic counseling is recommended.
Variant(s) of Uncertain Significance (VUS) Detected
Variant(s) of Uncertain Significance (VUS) are variants that do not fit into pathogenic, likely pathogenic, or benign classifications according to ACMGG and/or other relevant Professional standards, or the criteria for pathogenic or likely pathogenic and benign are contradictory for the variant. There is not enough information or conflict information at this time to support a more definitive classification of this variant on cancer risk.
Negative – No Reportable Variant(s) Detected
The result means that no pathogenic, likely pathogenic (LP) or variant of uncertain significance (VUS) were detected. Likely benign and benign variants are not included in the report and are available upon request.
Test Inconclusive or Test Failed
Test evaluation was inconclusive. Possible reasons are not enough DNA present in test specimen or the specimen failed quality assurance measures. A new specimen submission is recommended.
Variant Information Available
All variant information is available upon request. Vairants classified as benign and likely benign are not reported. Variants not detected by the performed assay may impact the phenotype.